The science¶
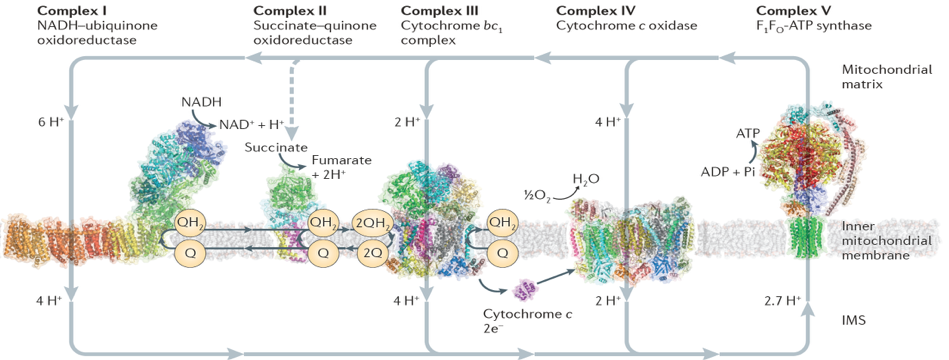
The focus of my current group is bioenergetics, specifically, mitonchondrial respiration. Efficient harvesting of solar energy will have far-reaching impact towards current energy needs. This makes bioenergetics the focus of modern energy research. Particularly, mitochondrial bioenergetics is receiving more importance than ever before. Mitochondria are the power-plant of most eukaryotic cells which employ electrons from NADH to synthesize ATP. The long-range goal of my research is to understand the mechanism of action of the respirasome consisting of membrane-embedded enzymes, named Complex I, III, IV and V.
Chemomechanical coupling of Complex I¶
Recent discovery of the atomic model of Complex I, the first and the largest protein complex in the mitochondrial respiratory chain, has provided a fantastic opportunity for such study. My current focus is on the chemomechanical coupling in Complex I, where electrons from NADH are utilized to reduce quinone while pumping protons across the membrane.
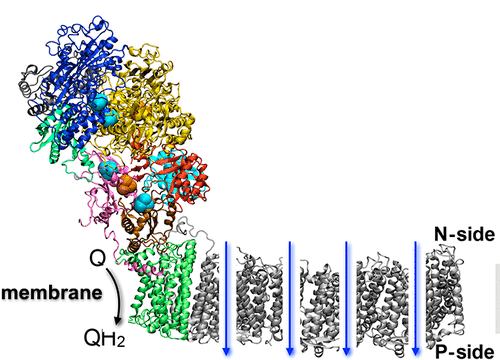
Complex I is an L-shaped protein with one soluble and one transmembrane arm. The transmembrane domain is responsible for pumping protons across the membrane. The soluble domain houses the binding sites for NADH as well as quinone. However, these two sites are far from each other, raising the obvious question, what couples the dynamics of the said binding sites? Other related questions are, how is the quinone traffick influenced by complex I reduction? Many of the answers are tied to chemomechanical coupling: mechanical motion of protein subunits arising out of chemical phenomenon (electron uptake). I have used large-scale MD simulations to understand the reduction-induced chemomechanical coupling in the soluble domain of complex I. By carefully defining the oxidation states of the 9 iron-sulfur (FeS) clusters of the enzyme, we identified the redox switches that couple FeS reduction with the quinone-binding site. Using free energy simulations, we demonstrated the design principles that govern quinol release. Finally, we showed how quinone-binding in the soluble domain couples proton transfer in the membrane domain --- through concerted, water-mediated hydrogen bond networks that are broken in the apo enzyme. Click here to read our JACS publication.
Proton transfer pathways in complex I¶
In the previous subsection, I talked about our computational studies on the soluble domain of complex I, including our initial observation that quinone binding was coupled to proton transfer in the membrane domain. Next, we took a closer look at the proton transfer mechanism of complex I. Complex I pumps four protons across the mitochondrial membrane using four proton channels. Unlike the first three channels that are closely related to antiporters, the fourth so-called “E-channel” is unique. In a collaborative work with the group of Prof. Marilyn Gunner at City University of New York, we showed that proton transfer through the E-channel is governed by a network of hydrogen bonded pathways. The Gunner group used Monte Carlo Continuum Electrostatics (MCCE) to sample protonation states, hydrogen orientation, and water occupancy of structures obtained from our MD simulations described above. Using this combined MD/MCCE approach, we identified clusters of complex I residues connected by residue-residue and residue-water-residue hydrogen bonded networks. Interestingly, this network was sensitive to the presence or absence of quinone in the soluble domain of complex I, further establishing our earlier finding that proton transfer is coupled to quinone binding. Click here to read our publication in BBA.
The methodology¶
The exciting science we do is made possible by advancements in computer hardware, parallel architecture, and recent advent of GPU-supported computations. Full exploitation of such developments can only happen through commensurate development of the existing simulation methodology. Our group as well as a few others are at the forefront of such method devlelopments targeted at pushing the limits of biomolecular simulation, both in terms of system size and simulation timescales that can be achieved realistically. The Singharoy group is a part of the team that develops and maintains NAMD, one of the most popular software suite for computational biophysics.
Data-driven simulations¶
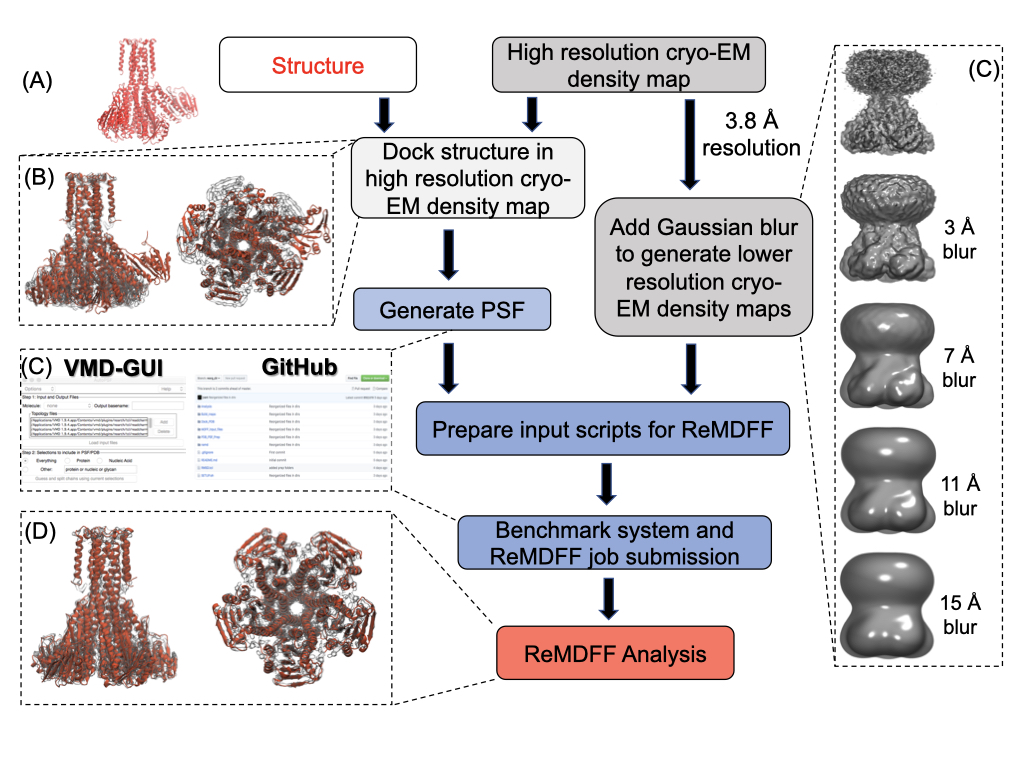
Cryo electron microscopy (cryo EM) is an emerging tool in structural biology, allowing protein structure determination at native-like environments. Compared to X-ray crystallography, cryo EM structures are often of poorer quality, necessitating the use of real-space refinement techniques for obtaining atomistic description of proteins. Molecular Dynamics Flexible Fitting (MDFF) is one such tool that has been highly successful on cryo EM data of resolution 5-7 Å or higher.
Cryo EM has undergone rapid advancement in recent times, routinely generating higher resolution ($<$5 Å) structures, which renders traditional MDFF insufficient. To circumvent this problem, resolution exchange MDFF (ReMDFF) was developed in 2016, which uses the concepts of replica exchange simulations in the resolution space for faster convergence. I have co-authored a book chapter which provides a detailed, step-by-step guideline for preparing, executing, and analyzing ReMDFF simulations, demonstrated by two case studies. Click here to view the chapter, published in "Protein Structure Prediction" issue of Methods in Molecular Biology
De novo protein folding¶
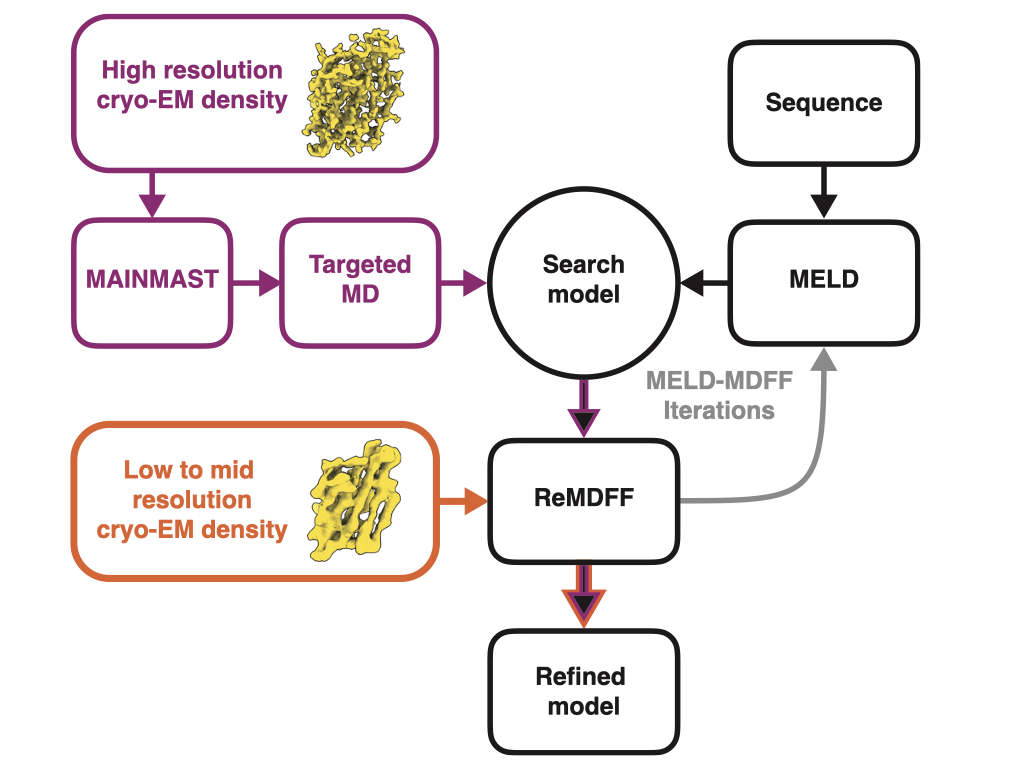
Multiple approaches exist for hybrid modeling, which combine MD simulation with experimental methods like NMR spectroscopy, X-ray crystallography and cryo EM. Each of these approaches use molecular replacement, where an initial search model is fitted to the experimental data. This has two consequences:
To overcome these problems, we have developed a protein modeling pipeline that interactively combines a minimal spanning tree based backbone tracing tool (MAINMAST), a bayesian-likelihood based protein-folding method (MELD), and a molecular dynamics fitting protocol (ReMDFF, referred to in the previous subsection). Primary sequence information and cryo EM images are the only input required, and images with resolution of as low as 5 Å are sufficient to get a structural model. Our method, which we call CryoFold, works well even with heterogeneous and sparse data. The software is automated and has a python/kivy-based graphical user interface. Click here to read our publication in Matter.